A new study led by C. Heshmatpour and J. Hauer from TU München studies exciton-exciton annihilation in a squaraine trimer. The experiment exploits 5th-order optical spectroscopy to study the evolution of the trimer after two-photon excitation into its bi-exciton state. Quantum chemistry computations performed by M. Menger, now located at Groningen, provide the required parameters to model the experimental signals within a Frenkel exciton model. The associated article Annihilation Dynamics of Molecular Excitons Measured at a Single Perturbative Excitation Energy just appeared in J. Phys. Chem. Lett.
Dynamics
Paper: Details in the surface hopping method
Our paper “Strong Influence of Decoherence Corrections and Momentum Rescaling in Surface Hopping Dynamics of Transition Metal Complexes” was just accepted in JCTC. In this work we investigated the reliability of the surface hopping method in the case of a transition metal complex described using a linear vibronic coupling model. We found that various seemingly unimportant parameters can have a strong influence on the results.
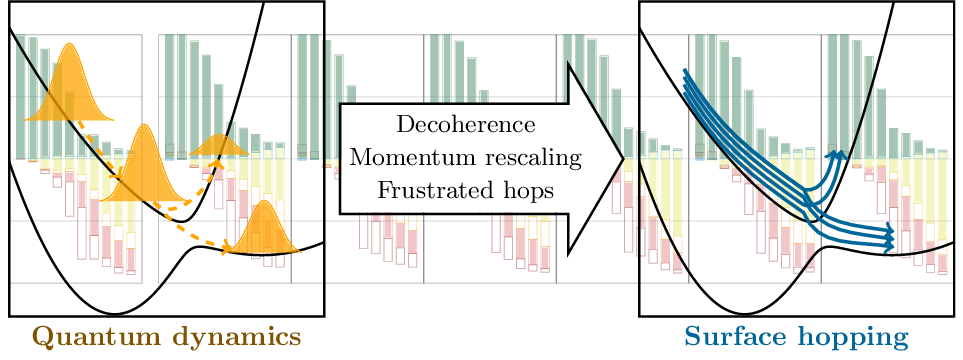
Preprint: Details in the surface hopping algorithm
Having discussed the influence of electronic structure methods in surface hopping dynamics in the last post and paper, we can now proceed to the surface hopping algorithm itself. To our surprise, algorithmic details such as the decoherence correction (energy-based decoherence or augmented FSSH), momentum rescaling and the treatment of frustrated hops can make a big difference. This is what we investigated in our new preprint “Strong Influence of Decoherence Corrections and Momentum Rescaling in Surface Hopping Dynamics of Transition Metal Complexes” available on ChemArxiv.
To have a well-defined reference, we used our new implementation of vibronic coupling models for surface hopping, which allows us to have a one-to-one comparison with accurate quantum dynamics computed at the MCTDH level of theory. As model system, we used a rhenium complex and studied its ultrafast intersystem crossing dynamics from the singlet to the triplet manifold following previous studies by our collaborators in Strasbourg [JCTC (2017), PCCP (2018)].
Paper: Electronic structure methods for dynamics simulations
The challenge about running photodynamics simulations is that the computational cost is often so high that one might have to compromise in terms of the electronic structure method used. One is tempted to just check the vertical excitations at one geometry and run the dynamics if those look alright. How this can go wrong is investigated in the paper “The Influence of the Electronic Structure Method on Intersystem Crossing Dynamics. The Case of Thioformaldehyde” that just appeared in JCTC. Take a look if you are interested.
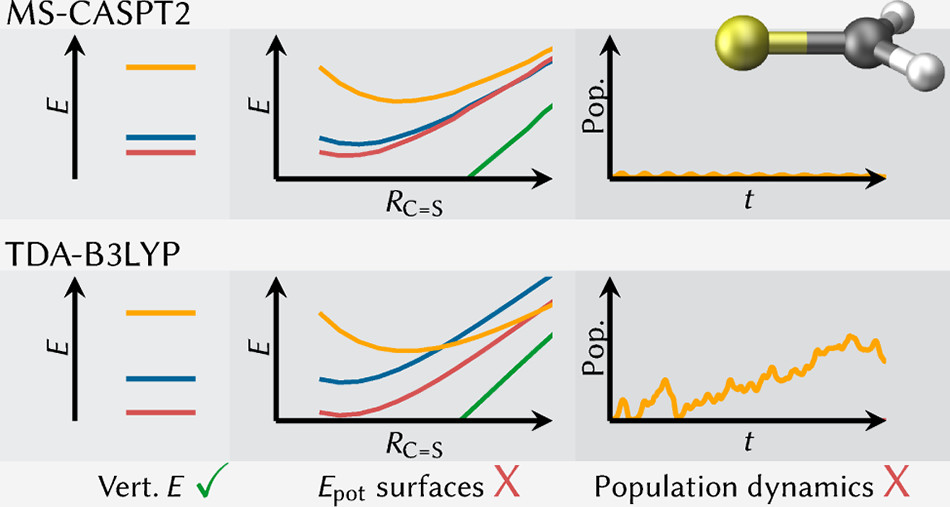
Copyright 2019 American Chemical Society.
Paper: Dynamics within an Exciton Model
Another paper working on improving the efficiency of surface hopping dynamics just appeared, this time in JCTC: “Surface hopping within an exciton picture – An electrostatic embedding scheme.” authored by M. F. S. J. Menger, F. Plasser, B. Mennucci, and L. González. In this paper, we explored the possibility of running nonadiabatic dynamics simulations within an exciton model. The main challenge in this endeavour was to derive a consistent energy expression for combining QM/MM electrostatic embedding calculations of the different chromophores.

To test the implementation, we ran simulations on a molecular dyad, where full TDDFT nonadiabatic dynamics simulations were available. Good agreement was found.
The method was implemented in the SHARC molecular dynamics package.
Paper: Highly-efficient photodynamics simulations
A new paper co-authored by F. Plasser just appeared in PCCP: “Highly efficient surface hopping dynamics using a linear vibronic coupling model.” The paper shows that it is possible to perform photodynamics simulations of nonadiabatic processes, such as internal conversion and intersystem crossing, at virtually no cost.
Book Chapter: Trajectory Surface Hopping
The book chapter “General Trajectory Surface Hopping Method for Ultrafast Nonadiabatic Dynamics” written by S. Mai, F. Plasser, P. Marquetand, and L. González of the book Attosecond Molecular Dynamics edited by M. J. J. Vrakking and F. Lepine just appeared online. Download and read it if you are interested in getting an introduction into nonadiabatic surface hopping dynamics.
Paper: Vibronic coupling constants
You can find our new paper “Interstate vibronic coupling constants between electronic excited states for complex molecules” that recently appeared in JCP. The purpose of this paper was the development of a method that allows to determine interstate vibronic coupling constants, which are a decisive ingredient for model Hamiltonians used in quantum dynamics. Our idea was to start with a method based on wavefunction overlaps that is commonly used for trajectory dynamics simulations and adapt it for the case of quantum dynamics.
Talk (March 20): Surface Hopping within an Exciton Picture
On March 20, at 2pm (room N1.12), Max Menger will give a seminar talk at the Materials Modelling Seminar about his work of performing photodynamics simulations of multichromohporic systems. The title of his talk will be: Surface Hopping Dynamics Simulations within an Exciton Picture. Please join if you are interested.