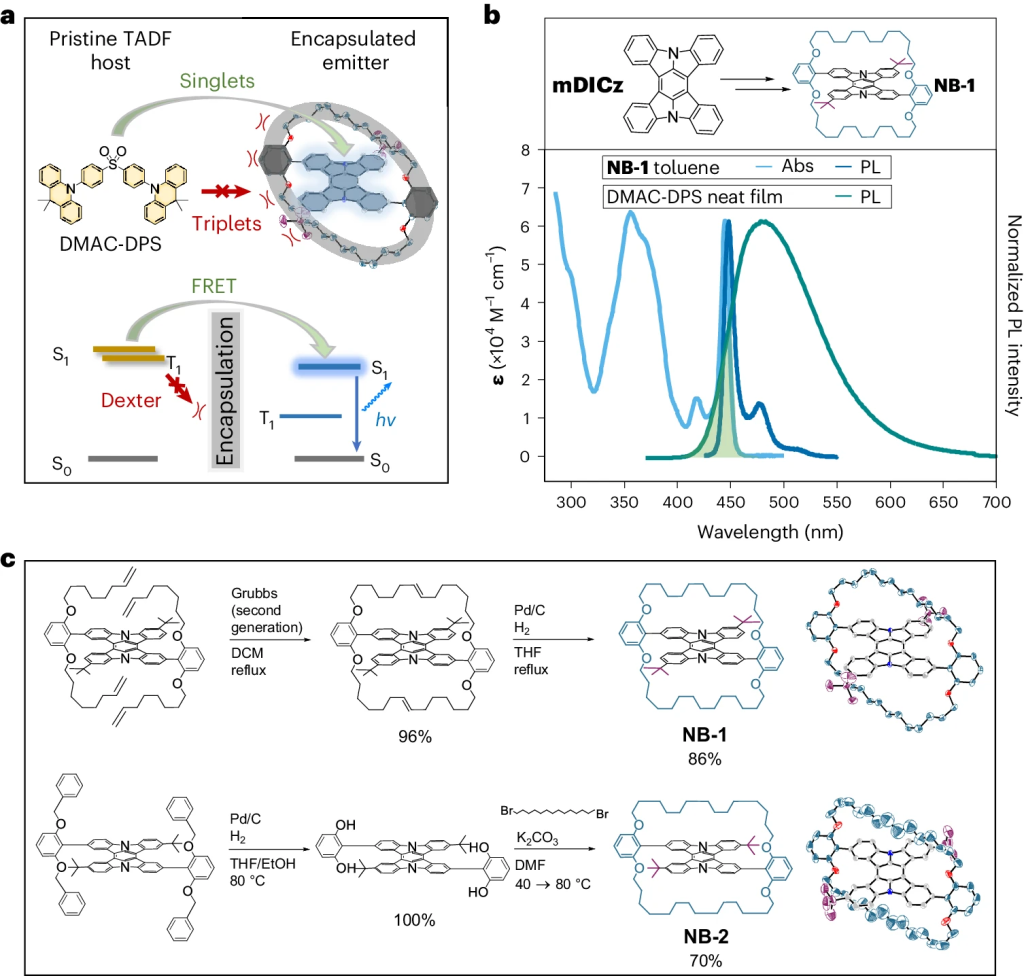
Hyperfluorescence is an emerging technique for generating highly efficient OLEDs by combining a triplet harvester with a bright emitter molecule. Current devices are overly complex due to the number of components involved hampering practical application. A new paper, led by Hugo Bronstein from the University of Cambridge presents an important step toward solving this problem. The idea is to encapsulate the emitter, thus, avoiding the need for a high-gap matrix. The approach is presented in the paper Suppression of Dexter transfer by covalent encapsulation for efficient matrix-free narrowband deep blue hyperfluorescent OLEDs, which just appeared in Nature Materials.