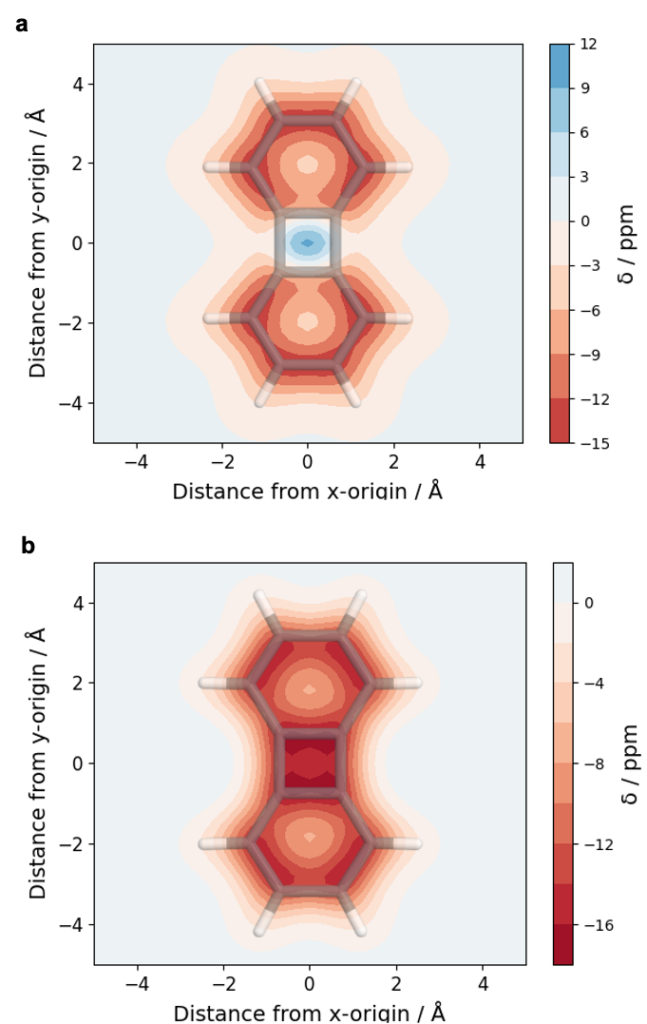
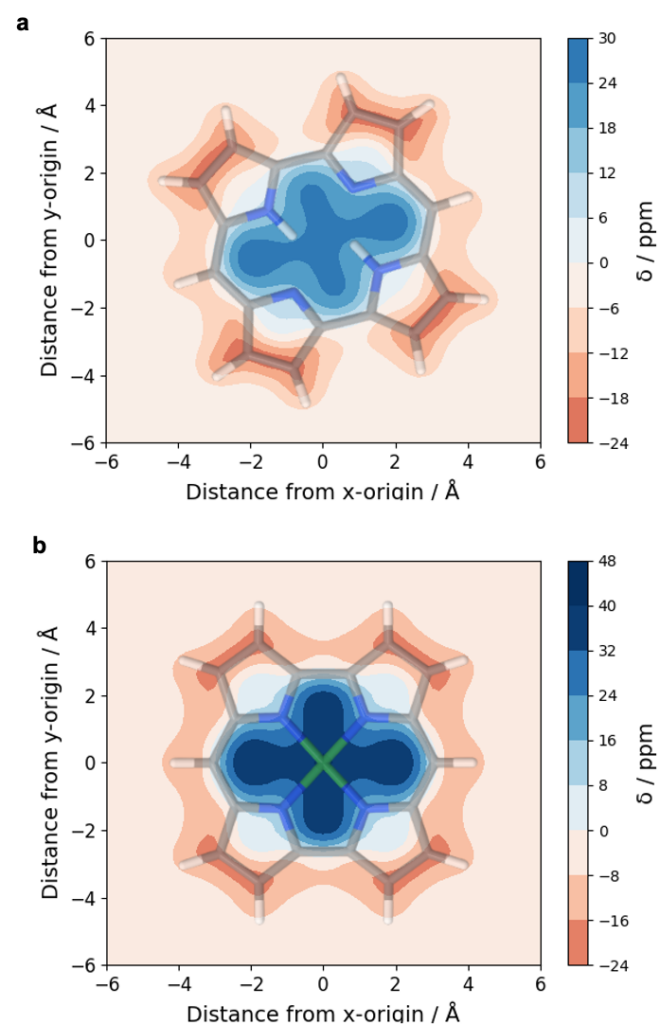
A recent JPCA article by Karadakov and Al-Yassiri highlights the differences in singlet and triplet aromaticity in naphthalene. To me this paper contains several striking observations:
- The singlet HOMO/LUMO transition (S2, 1La) is shown to be strongly aromatic whereas the triplet HOMO/LUMO transition (T1, 3La) is antiaromatic. Does this mean states reached by the same kind of orbital transition behave differently depending on their spin-multiplicity?
- The aromatic S2 lies above the antiaromatic S1 even though S2 is the HOMO/LUMO transition. Does this mean that singlet antiaromaticity is actually a stabilising effect?
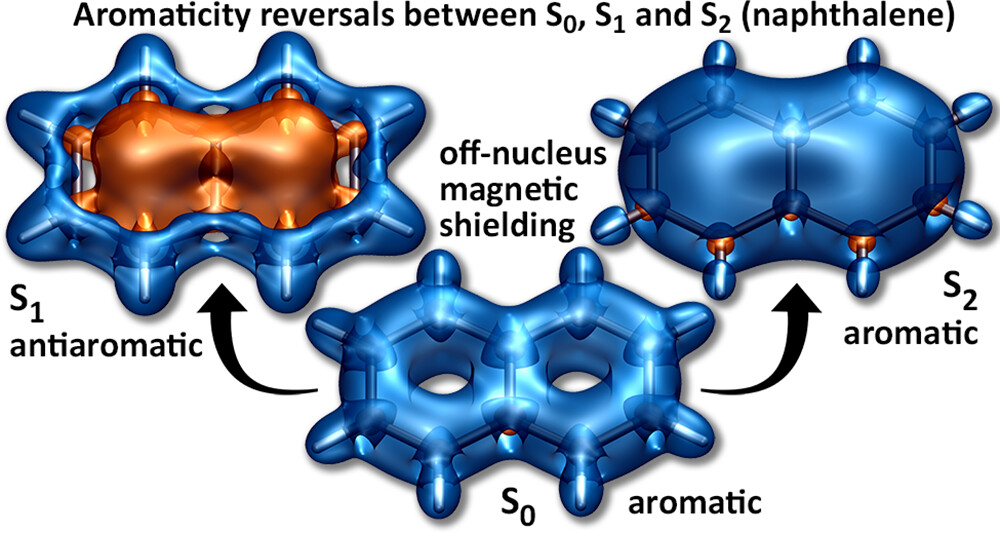
We have discussed the excited states of naphthalene from an entirely different viewpoint in a recent J. Chem. Theory Comput. article. It would be fascinating to combine the two viewpoints.