Our new paper Quantification of the Ionic Character of Multiconfigurational Wave Functions: The Qat Diagnostic just appeared in the Journal of Physical Chemistry A.
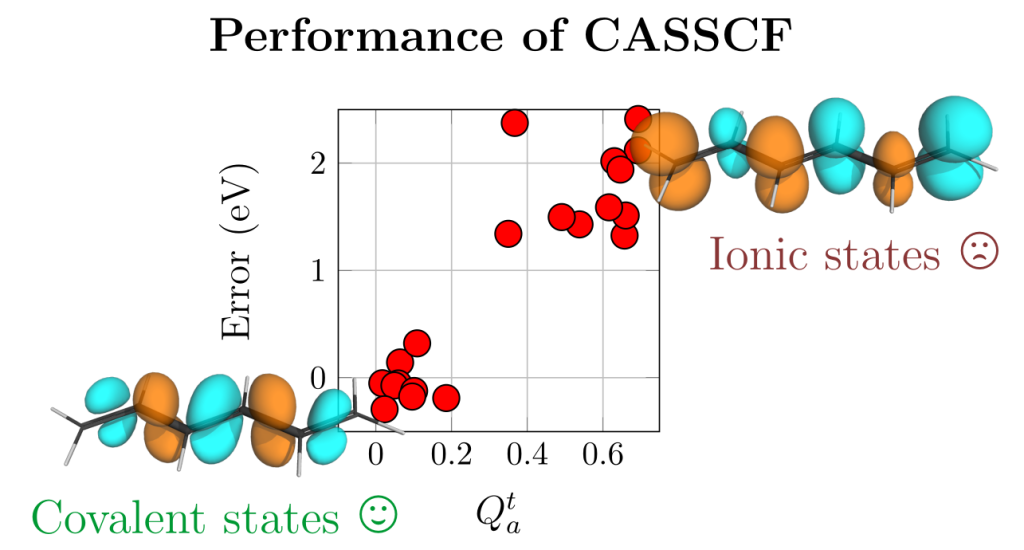
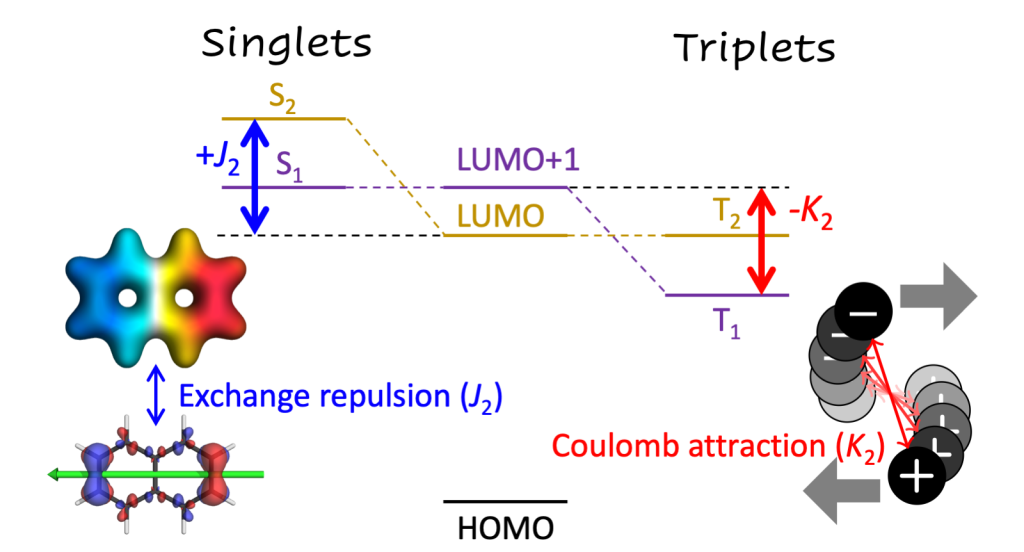
The paper deals with the fact that the widely used CASSCF method, if not used carefully, can yield large errors (1-2 eV) in vertical excitation energies. This problem arises for ionic states, as defined within valence bond theory. Within this work we developed a simple diagnostic to identify ionic states. We found a good correlation between the new diagnostic (Qta) and the error, as shown in the figure above.
We hope that the new diagnostic will be useful similar to analogous diagnostics identifying charge transfer states in TDDFT computations. This will give users the possibility to spot potential problems quickly.
On-going work is concerned with going from just diagnosing the problem to developing a numerical correction term to fix the problem.