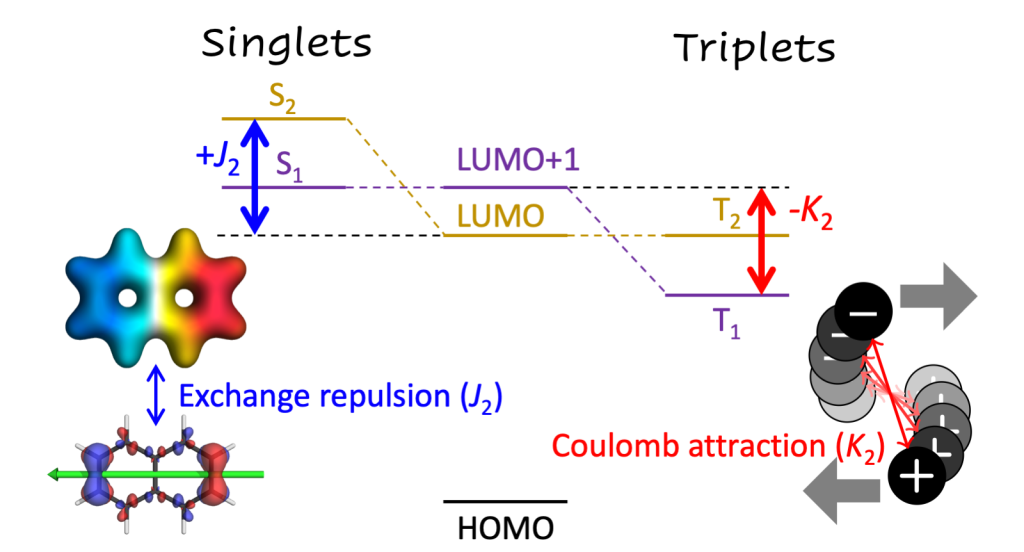
We just posted a preprint discussing a question I have been wondering about for a while: Why is the lowest excited state of a molecule not always the HOMO/LUMO transition? More generally we show how singlet and triplet state energies are affected in different ways by post-MO energy terms.
How do macrocycles with [4n] electrons behave? Are there signatures of their formal antiaromaticity and how can their properties be tuned for practical applications? A recent study, led by Florian Glöcklhofer (Imperial College, London) endeavours to tackle these questions. A set of macrocycles based on [2.2.2.2]cyclophanetetraenes was synthesised, their redox and optical properties were measured, and a detailed computational analysis was performed.
Clear signatures of the unique properties of these macrocycles was found considering their large Stokes shifts (>1.5 eV) along with the ease of producing doubly charged states. A detailed computational analysis traces these properties back to the aromaticity of the excited and doubly charged states, respectively. In addition, it is illustrated how the properties of the macrocycles can be systematically varied with introduction of functional groups and variation of the aromatic units.
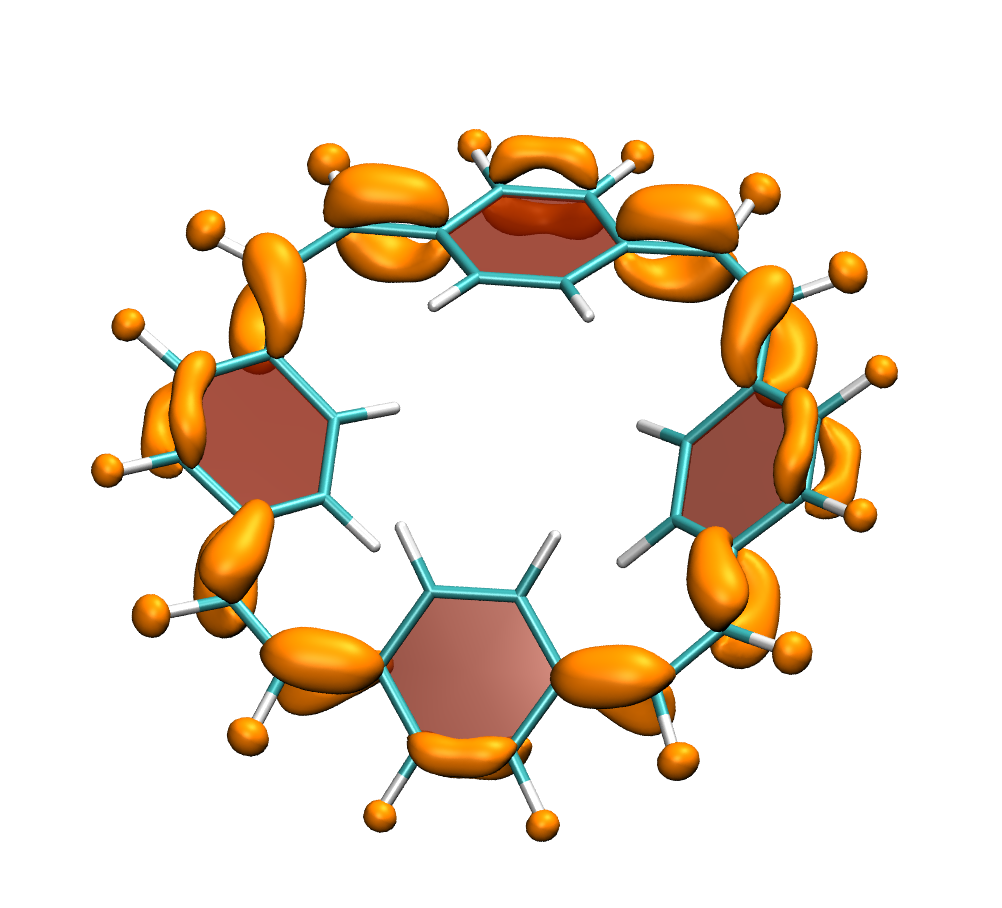
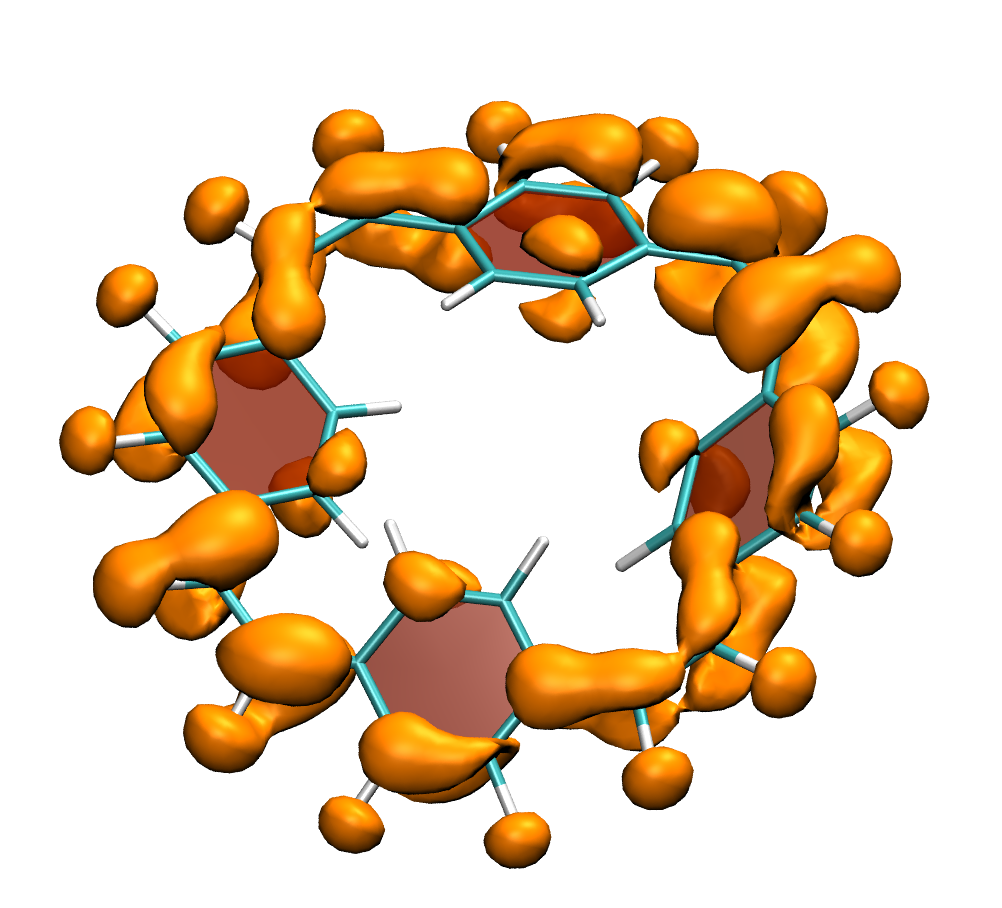
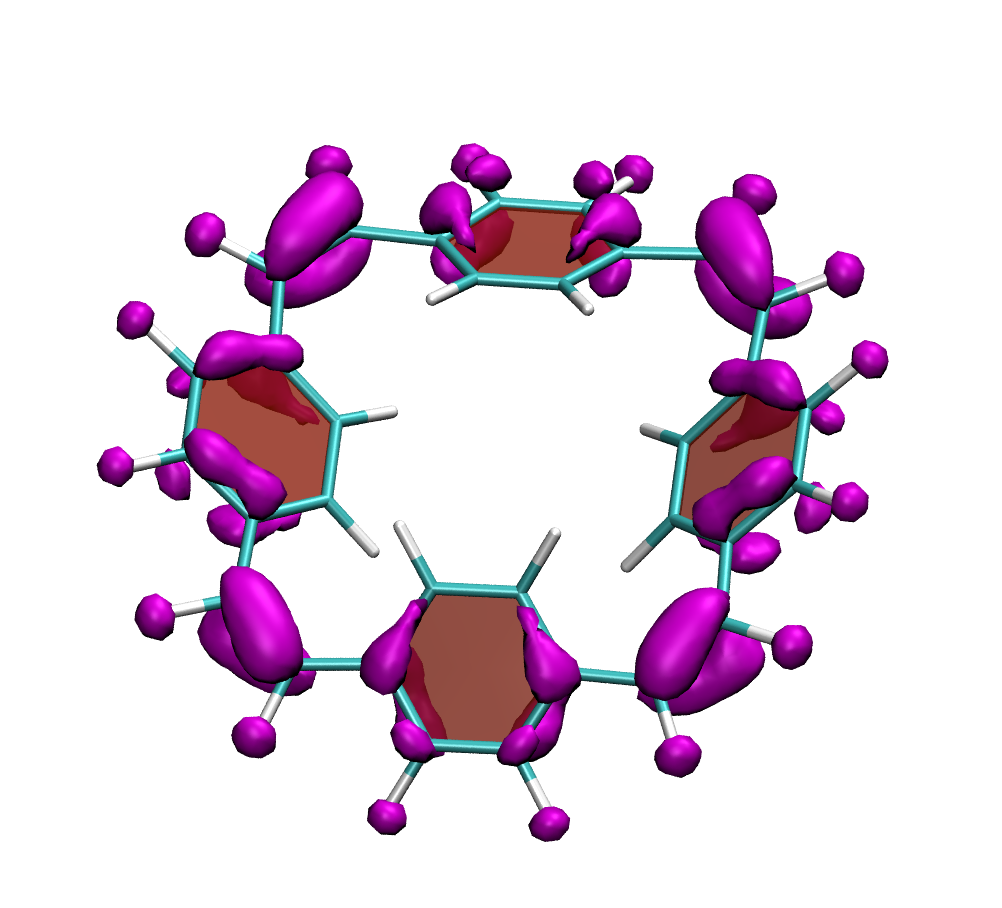
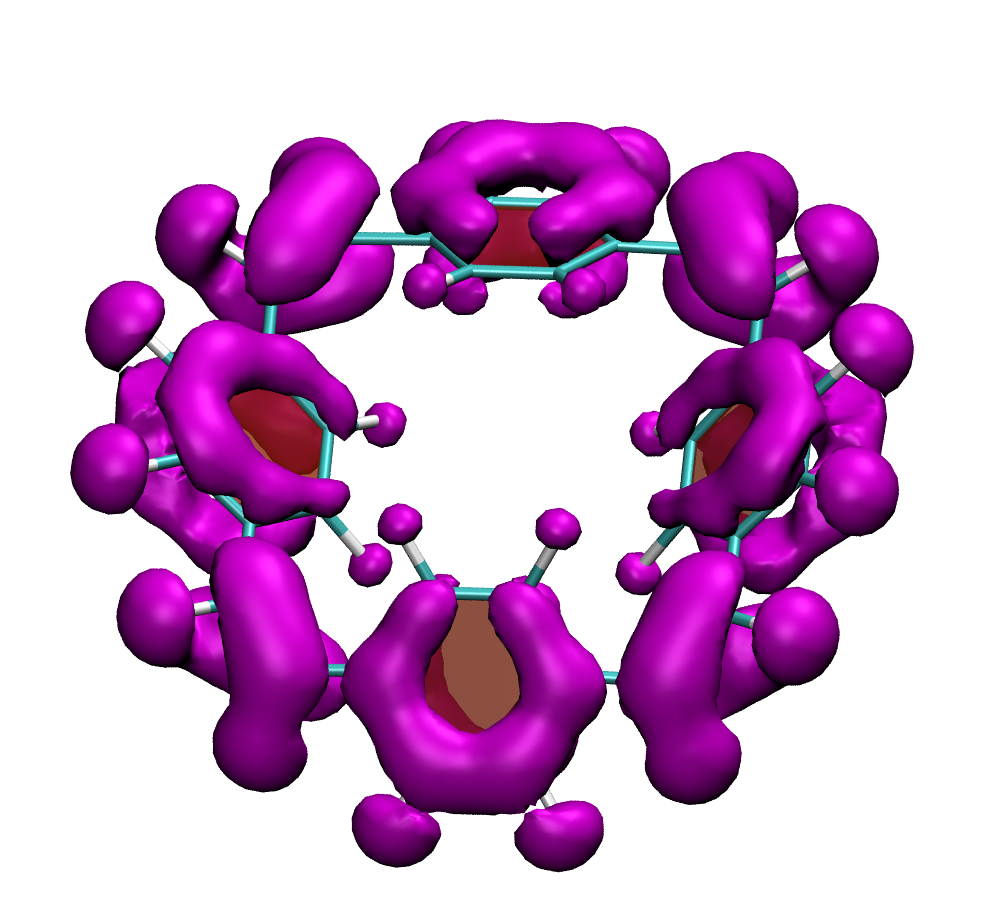
Below, electron density difference plots for the charged states of the parent molecule paracyclophanetetraene are shown highlighting the cyclic symmetry of the electron attachment. The 2+/2- and 6+/6- states are aromatic whereas the 4+/4- singlet states are antiaromatic.
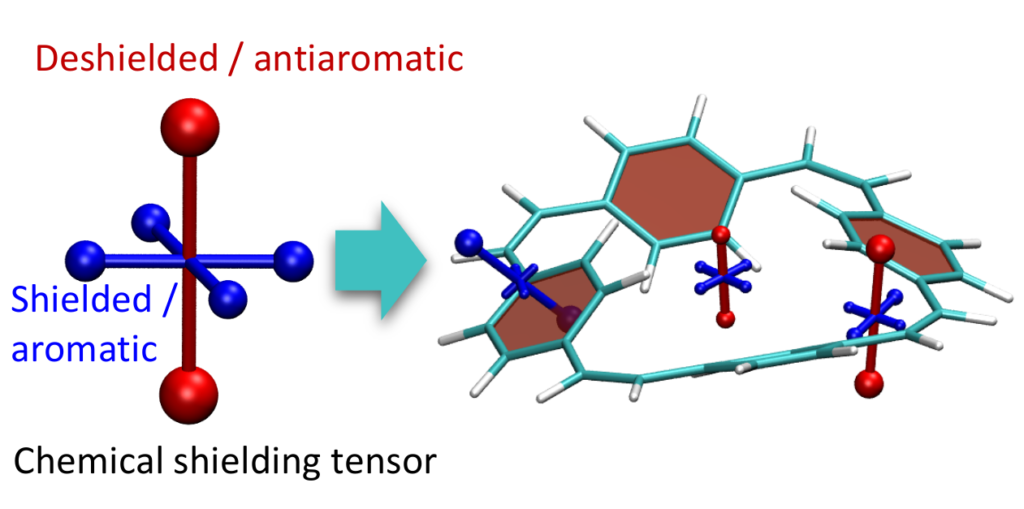
Aromaticity is a ubiquitous yet elusive concept in chemistry and chemists have spent a great deal of effort on developing methods to quantify and visualise aromaticity. One particularly popular method is the nucleus independent shift (NICS), which can be seen as a virtual NMR experiment carried out within a conjugated ring to evaluate the enhanced chemical shielding induced by aromatic ring-currents. Strikingly NICS also allows to quantify antiaromaticity, as this induces a net deshielding effect within the ring. NICS provides a powerful quantitative aromaticity criterion but the main challenge for its graphical representation is that the chemical shielding is a 3×3 tensor, which is difficult to visualise with the existing methods.
Therefore, we have developed a new method for the visualisation of chemical shielding tensors (VIST), which provides a local representation of the shielding tensor along with the molecular structure. The method, thus, allows to probe local aromaticity along with the underlying anisotropy of the shielding. The method is described in the preprint “3D Visualisation of chemical shielding tensors to elucidate aromaticity and antiaromaticity” available on ChemRxiv.
Within the preprent we exemplify the main concepts in the benzene and phenanthrene molecules and continue by studying
the interplay of ground state antiaromaticity and Baird triplet state aromaticity in the potential singlet fission chromophore cyclobuta[l]phenanthrene,
local aromaticity in the neutral formally antiaromatic ground state along with global aromaticity in the doubly reduced state of paaracyclophanetetraene,
Second, a more extensive paper exploring how far we can use information from excited-state wavefunction analysis tools to understand excitation energies beyond the molecular orbital picture. The energy of a correlated electron-hole pair is derived using diagrammatic techniques and this information is further used for a graphical depiction in terms of different charge distributions and their electrostatic potentials. Doing so turned out not as easy as hoped for but was very exciting. Find more here: Toward an Understanding of Electronic Excitation Energies Beyond the Molecular Orbital Picture by P. Kimber and F. Plasser.
To have a well-defined reference, we used our new implementation of vibronic coupling models for surface hopping, which allows us to have a one-to-one comparison with accurate quantum dynamics computed at the MCTDH level of theory. As model system, we used a rhenium complex and studied its ultrafast intersystem crossing dynamics from the singlet to the triplet manifold following previous studies by our collaborators in Strasbourg [JCTC (2017), PCCP (2018)].
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy