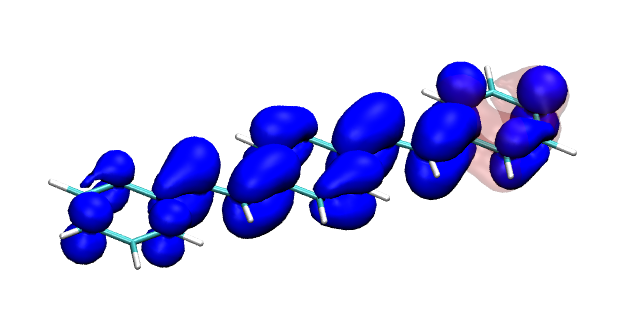
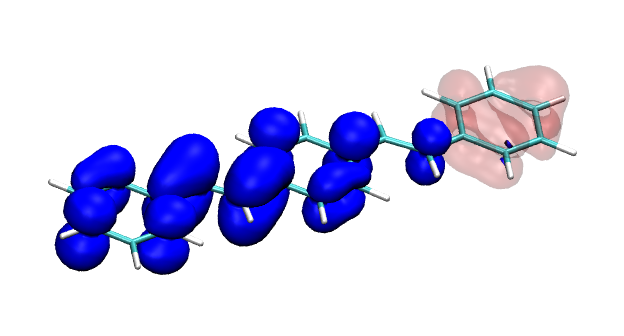
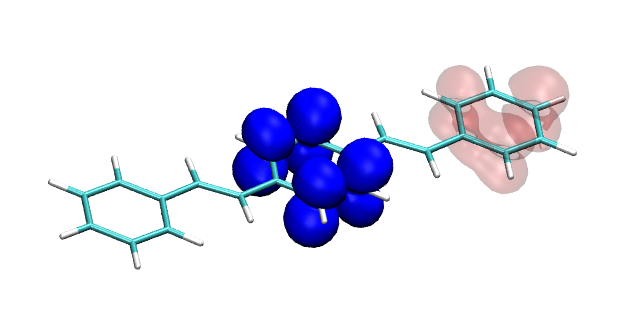
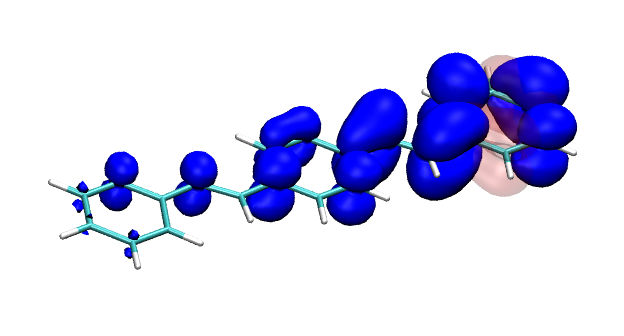
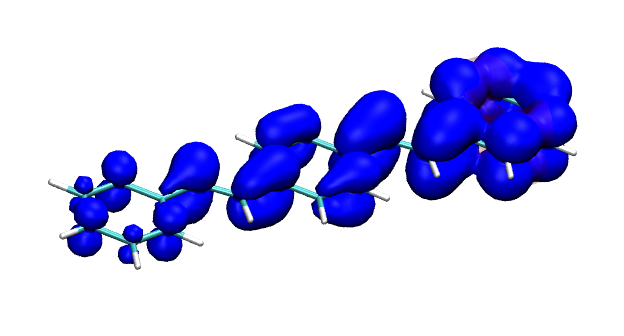
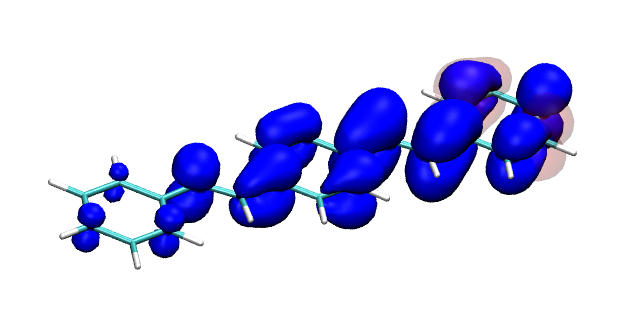
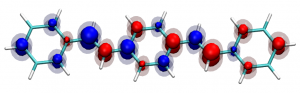
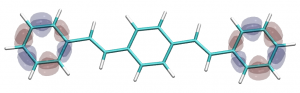
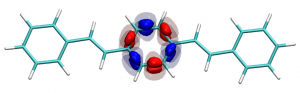
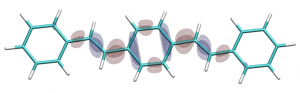
You can find the new paper describing the OpenMolcas package in JCTC – OpenMolcas: From source code to insight. OpenMolcas represents the open-source release of the previously commercially distributed Molcas package. Use OpenMolcas to gain access to powerful multireference methods for free and to have full control if you need to modify the source.
My own contributions to Molcas are concerned with the implementation of the wavefunction analysis module &WFA , as described in JCTC 13, 5343 (2017), and the interface to Columbus. Let me know if you have any questions about these.