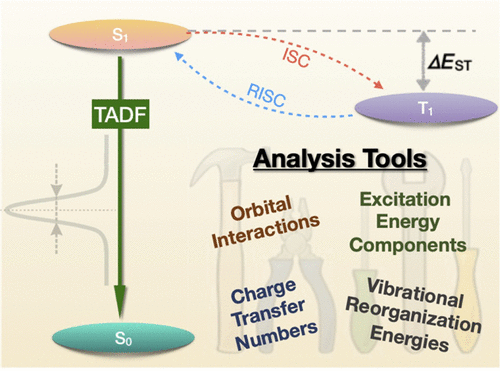
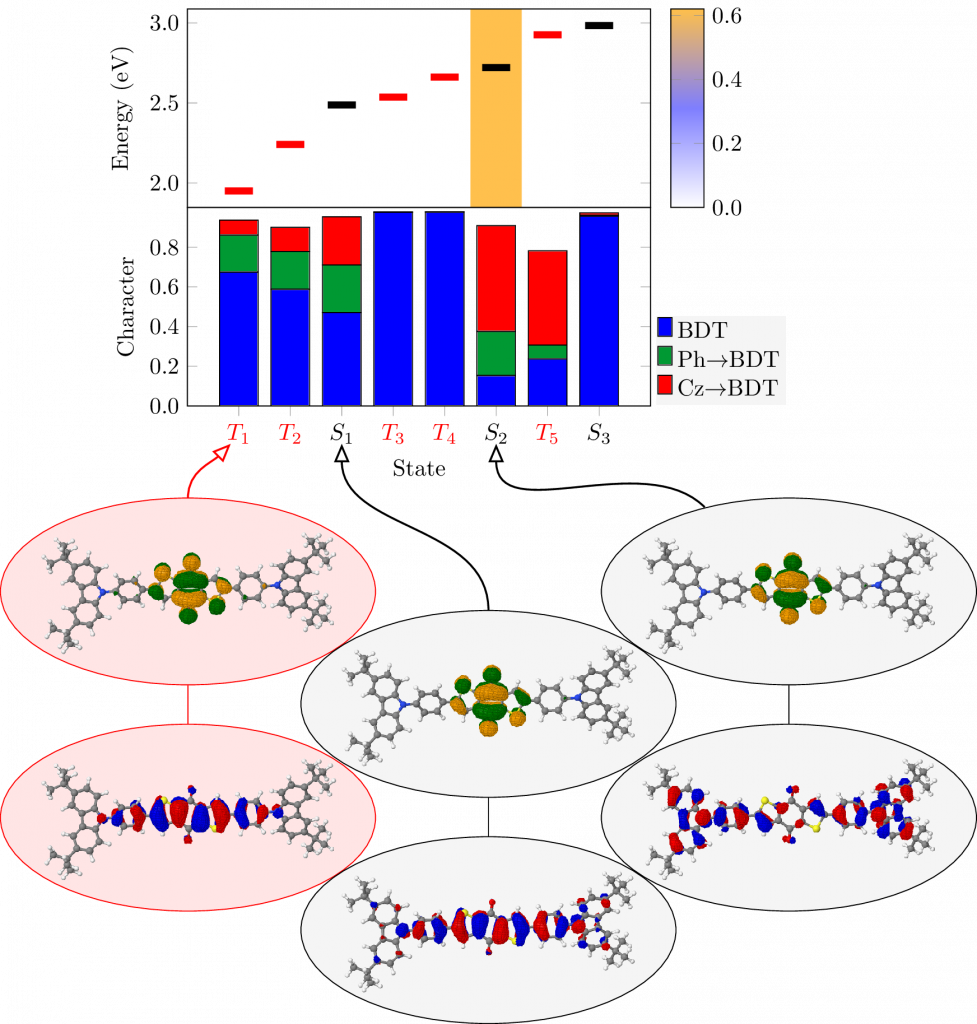
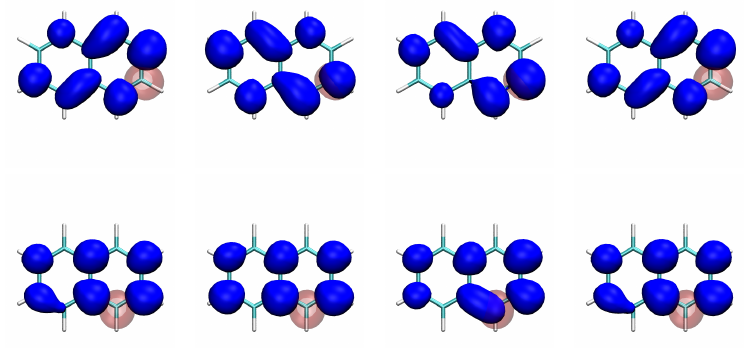
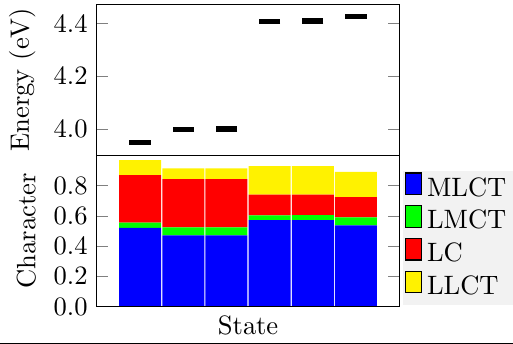
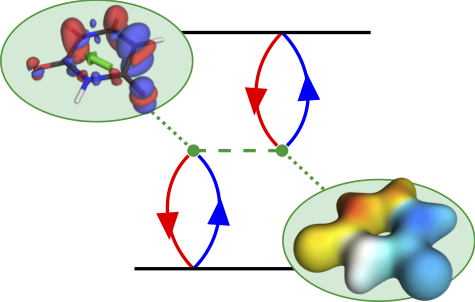
Thermally activated delayed fluoresence (TADF) is an exciting modern research area aimed at producing new OLED emitters. From a theoretical perspective TADF is particularly fascinating because it requires a detailed understanding of the different terms that contribute to the singlet and triplet excitation energies of the molecules studied. In a recent study led by Yihan Shao from the University of Oklahoma, we investigated a recently developed TADF emitter and showed how a combination of different wavefunction analysis tools provides deep insight into its excited-state properties. The paper just appeared in J. Phys. Chem. Lett.: Elucidating the Electronic Structure of a Delayed Fluorescence Emitter via Orbital Interactions, Excitation Energy Components, Charge-Transfer Numbers, and Vibrational Reorganization Energies.