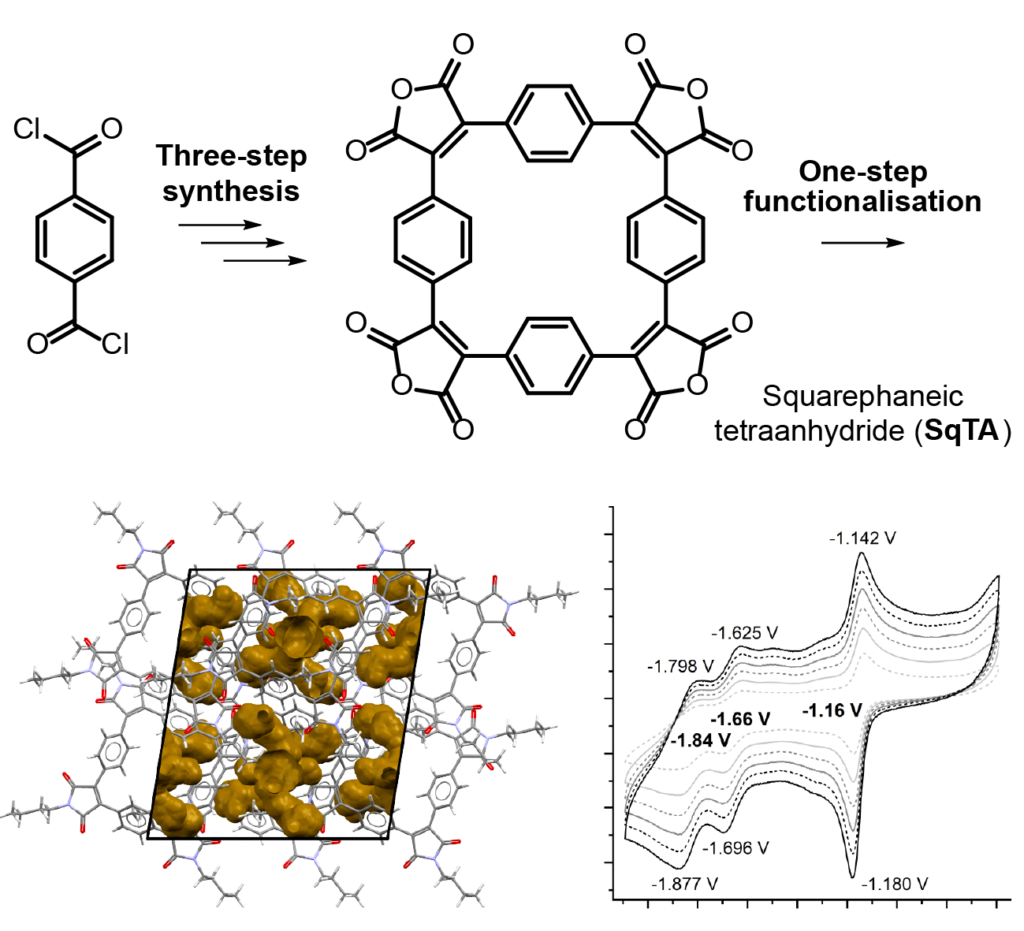
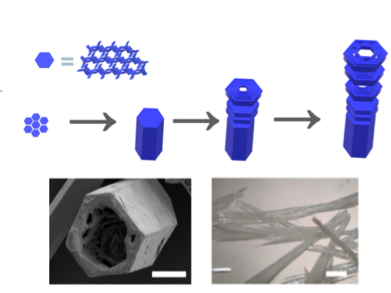
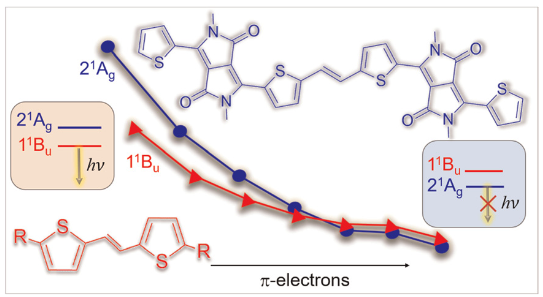
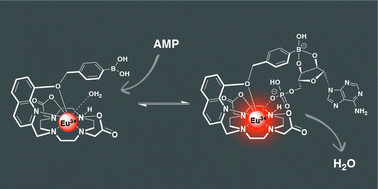
Our new paper “Classification of Doubly Excited Molecular Electronic States” just appeared in Chemical Science.
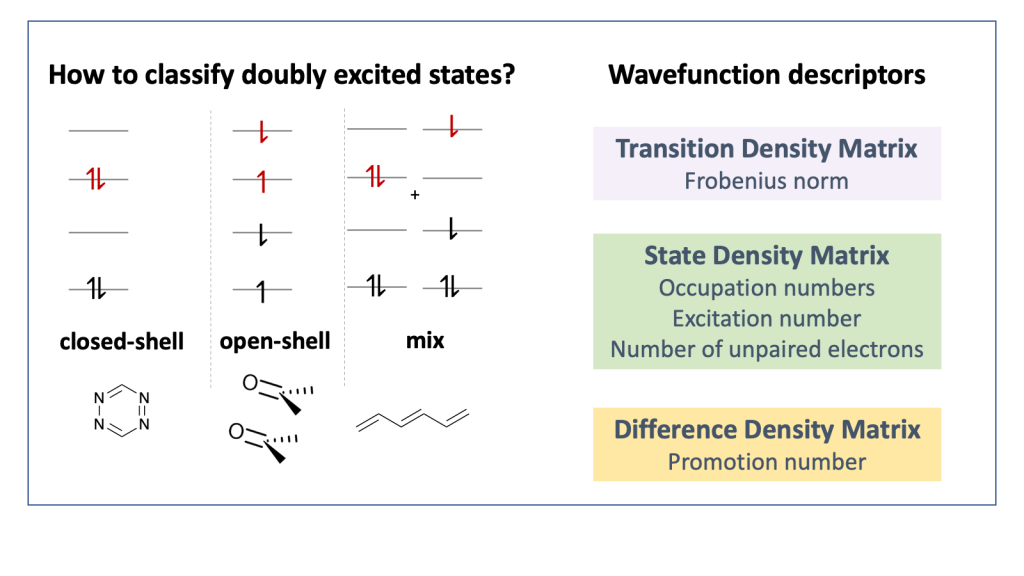
The topic of doubly excited states has been discussed quite controversially in the literature over the last couple of years, see for example JACS, 139, 13770 (2017) and JCTC 14, 9 (2018), and it is often disputed whether to classify a state as doubly excited at all. To contribute to this discussion we worked on the development of a physically motivated definition of doubly excited character based on operator expectation values and density matrices, which works independently of the underlying orbital representation. We hope that this approach will provide new understanding on these issues.