Today Felix is giving a talk at the Computational PhotoChemistry Online Meeting 2021: Tuning photophysical properties via excited-state aromaticity
The talk discusses the effects of aromaticity on excited-state energies and other properties. Strategies for quantifying and visualising aromaticity are shown as well.
Three recent papers are discussed:
A method for the visualisation of chemical shielding tensors (VIST) [EJOC 2021, 17, 2529]
How do macrocycles with [4n] electrons behave? Are there signatures of their formal antiaromaticity and how can their properties be tuned for practical applications? A recent study, led by Florian Glöcklhofer (Imperial College, London) endeavours to tackle these questions. A set of macrocycles based on [2.2.2.2]cyclophanetetraenes was synthesised, their redox and optical properties were measured, and a detailed computational analysis was performed.
Clear signatures of the unique properties of these macrocycles was found considering their large Stokes shifts (>1.5 eV) along with the ease of producing doubly charged states. A detailed computational analysis traces these properties back to the aromaticity of the excited and doubly charged states, respectively. In addition, it is illustrated how the properties of the macrocycles can be systematically varied with introduction of functional groups and variation of the aromatic units.
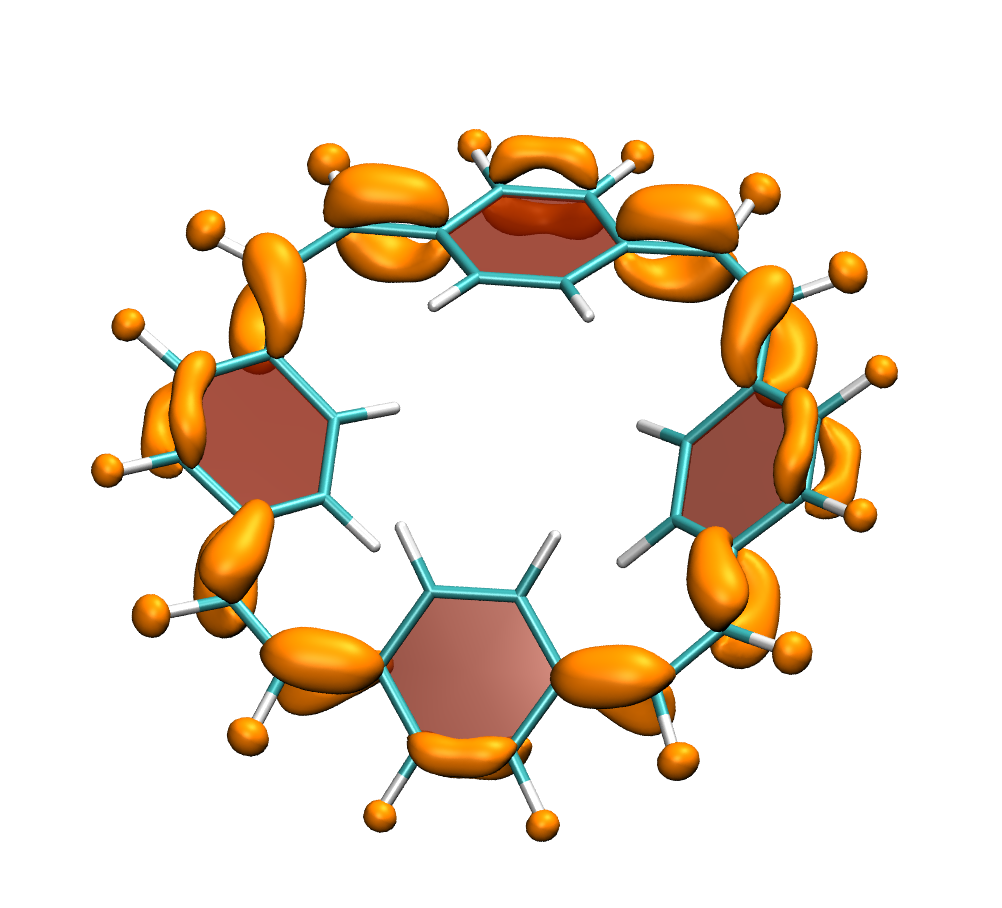
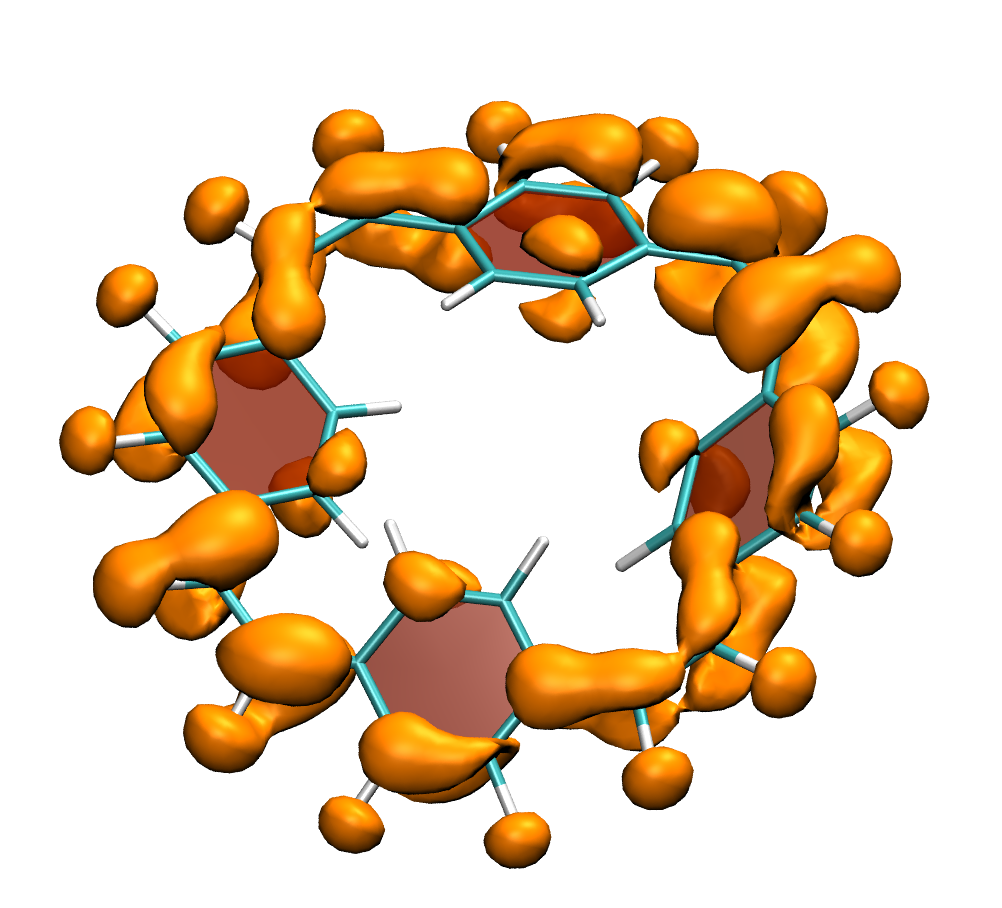
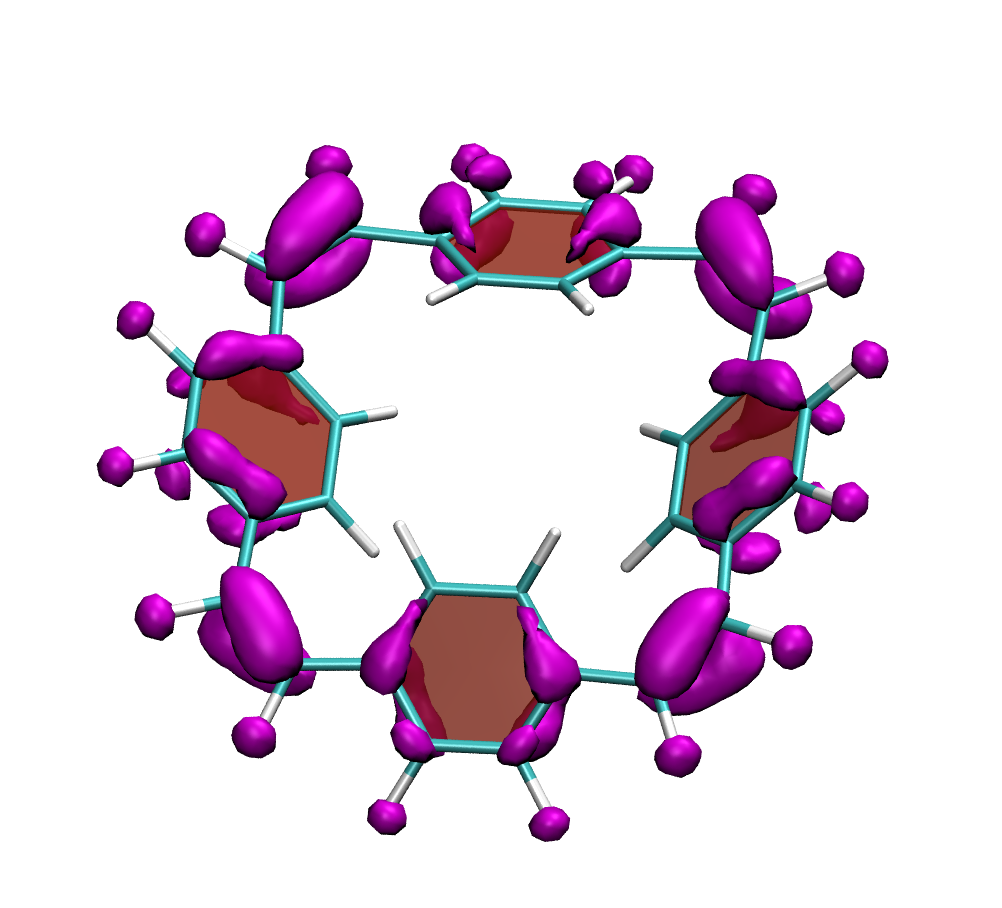
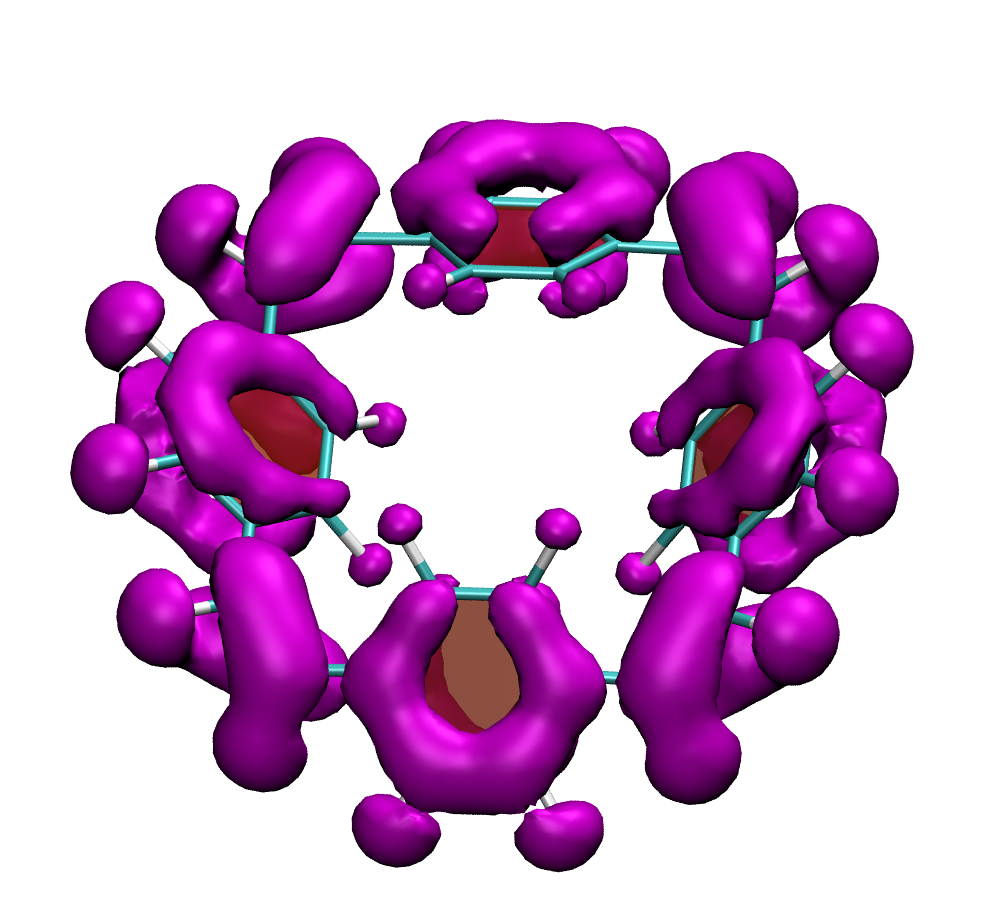
Below, electron density difference plots for the charged states of the parent molecule paracyclophanetetraene are shown highlighting the cyclic symmetry of the electron attachment. The 2+/2- and 6+/6- states are aromatic whereas the 4+/4- singlet states are antiaromatic.
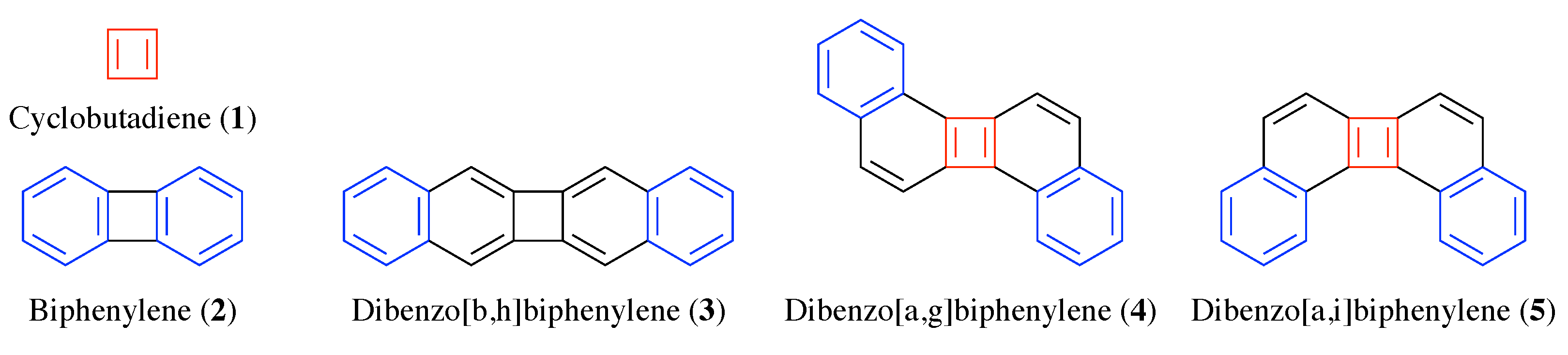
Can you sort these molecules according to increasing triplet excitation energies?
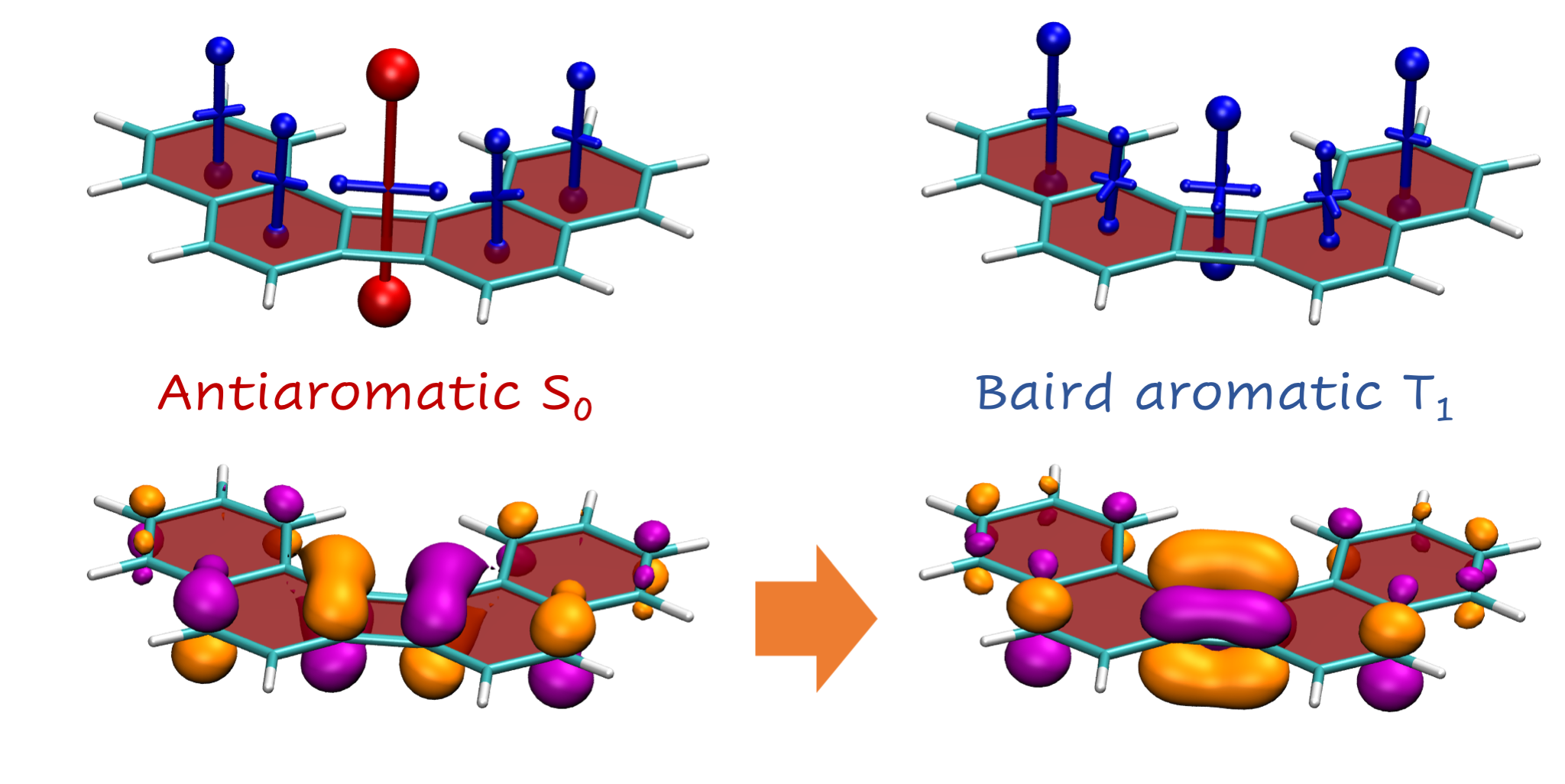
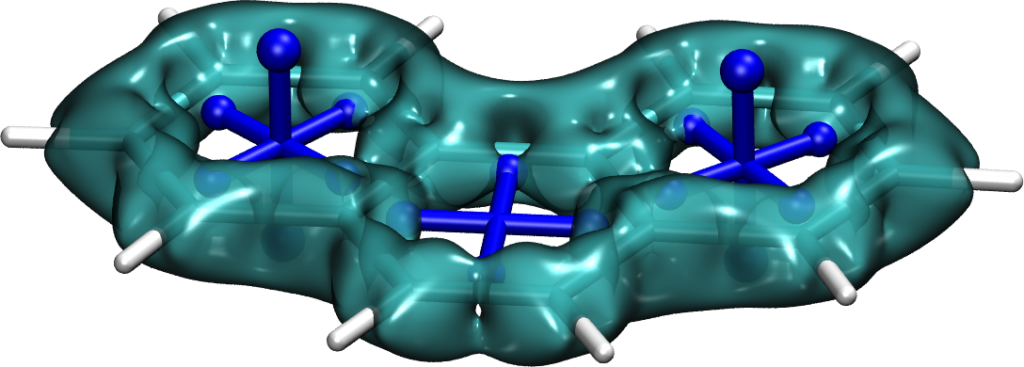
Some basic considerations might suggest that energies go down as the size of the molecule increases. But this is incorrect. The decisive feature of these molecules is their ground-state antiaromaticity along with their potential for excited-state Baird aromaticity. Triplet excitation energies increase sharply going from 1 (0.1 eV) via 2 (1.9 eV) to 3 (2.6 eV). This can be understood in the sense that antiaromaticity is blurred as the molecule becomes larger.
More strikingly, when going from 3 to 4 or 5, the energy drops again dramatically down to 1.0 eV. This effect is explained following Ayub et al. by the simple fact that these molecule possess resonance structures with simultaneous quartets and sextets.
A new study led by J. Lachner from the Helmholtz-Zentrum Dresden describes a method for detecting 26Al via Ion-Laser Interaction Mass Spectrometry using a particle accelerator.
Quantum chemical calculations highlight the different energetics of 26MgO and 26AlO, which are separated with high specificity despite being isobars.
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy