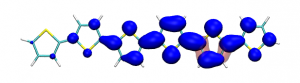
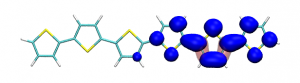
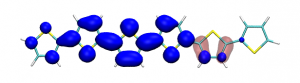
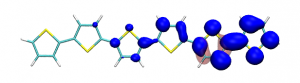
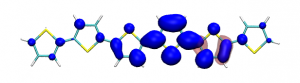
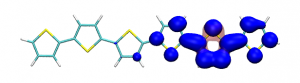
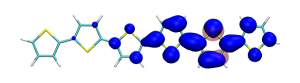
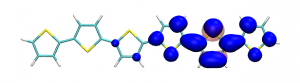
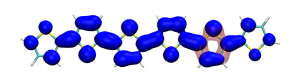
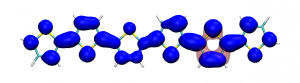
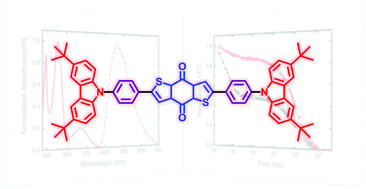
Small modifications can make a big difference. Swapping anthraquinone for a thiophene based acceptor in a donor-acceptor-donor system produces the desired red shift in the emission but limits its quantum efficiency. This conclusion was drawn from a recent paper lead by Stephanie Montanaro and Iain Wright at Loughborough: Red-shifted delayed fluorescence at the expense of photoluminescence quantum efficiency – an intramolecular charge-transfer molecule based on a benzodithiophene-4,8-dione acceptor which just a appeared in PCCP.
Computations reveal that the reason for the reduced emission quantum efficiency lies in the presence of low-lying locally excited states (4 triplets and one singlet) on the central unit.