Our new paper “Impact of Varying the Phenylboronic Acid Position in Macrocyclic Eu(III) Complexes on the Recognition of Adenosine Monophosphate“, led by S. E. Bodman and S. J. Butler from Loughborough, just appeared in Organic Chemistry Frontiers. The paper is the second in a series studying the anion sensing properties of Eu(III) complexes with phenylboronic acids.
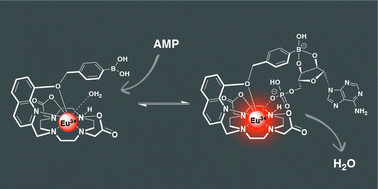
Aside from reporting the synthesis and anion binding, the paper presents new strategies for the computational analysis of such complexes. Aside from modelling the geometries by density functional theory, high-level multireference methods in OpenMolcas were applied to study the luminescence properties. These first principles computations offer a promising approach to access the emission spectra of lanthanide complexes, aiding the design of responsive lanthanide probes with specific photophysical properties